Май
Стабилизация пиролизного масла с помощью каталитического гидрообессоливания
1.Введение
Биомасса — единственный устойчивый источник углерода, следовательно, единственная альтернатива нефти и её производным. Исследования по применению биомассы для получения биотоплива первого поколения быстро развиваются (например, биоэтанол из сахаров и крахмалов, биодизель из растительных масел). Однако лигноцеллюлозную биомассу трудно напрямую преобразовать в транспортное топливо.
Текущие исследования сосредоточены на косвенных методах:
i) Фракционирование биомассы и ферментация целлюлозных и гемицеллюлозных фракций в этанол
ii) Газификация всей биомассы для получения синтез-газа с последующим превращением, например, в метанол или синтетический дизель по Фишеру-Тропшу.
Хотя экономически выгодны крупные масштабы переработки (до 100 т/ч в пересчёте на сырую нефть), это создаёт сложности с биомассой, так как она рассредоточена, а её сбор дорогостоящий. Более того, различные виды биомассы сильно различаются по структуре и составу, имеют низкую энергоёмкость по сравнению с ископаемым сырьём и часто содержат много влаги и золы.
Эти недостатки можно преодолеть, если предварительно переработать биомассу локально, увеличить её плотность (2–10 т/ч) и желательно одновременно «очистить». Полученный промежуточный продукт можно транспортировать в крупную центральную установку, где он будет переработан в финальный продукт (масштабом 50–200 т/ч). Одной из перспективных технологий для этого считается быстрый пиролиз (см. обзор Venderbosch & Prins, 2010).
Пиролизные жидкости содержат незначительное количество золы и обладают объёмной энергетической плотностью, в 5–20 раз превышающей таковую у исходной биомассы. Однако такое масло имеет кислую природу, является полярным и не смешивается с обычной нефтью. Кроме того, оно нестабильно: (ре)полимеризация органических веществ, содержащихся в масле, приводит со временем к увеличению вязкости.
Таким образом, пиролизное масло не следует рассматривать как традиционное «нефть» — это скорее сироп, богатый углеводами. Подтверждением этому служит метод фракционирования с помощью растворителей: основная часть масла — до 70% — легко извлекается водой. Типичный пример показан на рисунке 1 (и проанализирован в таблице 1), где пиролизное масло разделено на несколько групп соединений, различающихся по содержанию кислорода и молекулярному размеру (Oasmaa и др., 2003).
Интересно, что большая фракция, нерастворимая в эфире, содержит значительное количество соединений, подобных сахарам, и имеет внешний вид, напоминающий сироп. Сообщается, что высокая концентрация кислорода в пиролизном масле является основной причиной его нестабильности. Однако, вероятно, дело не столько в самом кислороде, сколько в характере и реакционной способности содержащих кислород функциональных химических групп. Особенно карбонильные соединения (альдегиды и кетоны) представляются ответственными за термическую нестабильность, и их превращение в менее реакционноспособные органические группы (например, в соответствующие спирты) кажется вполне жизнеспособным решением.
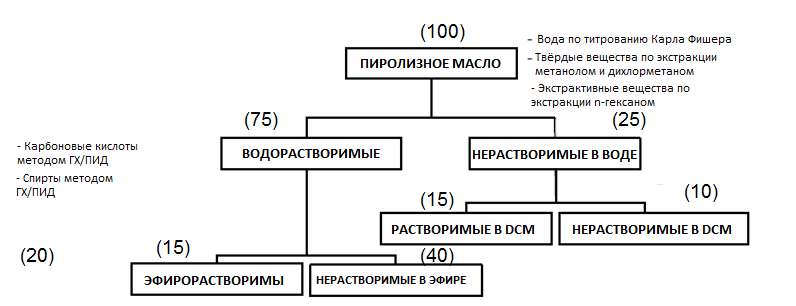
Рисунок 1.
Схема фракционная (Oasmaa, 2003)
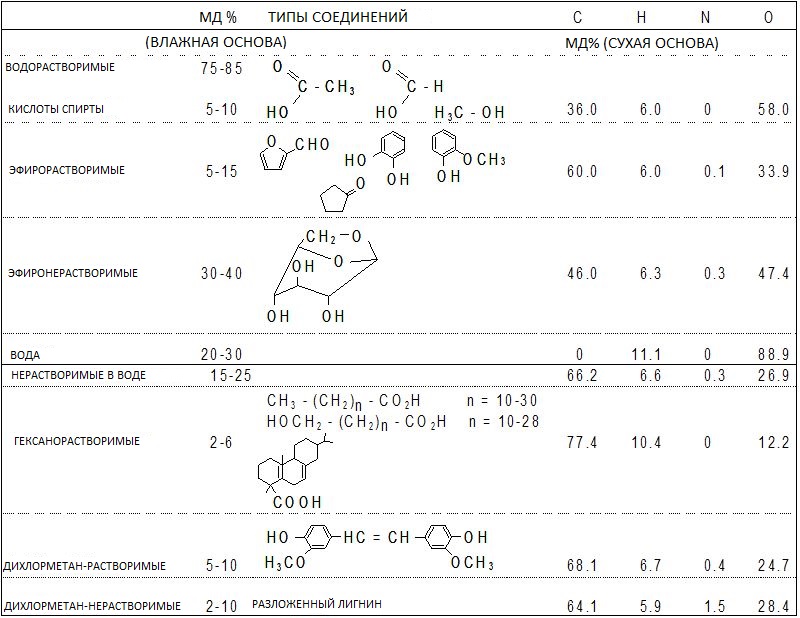
Стол 1.
Химический состав эталонного масла из древесины сосны и его фракций (Oasmaa, 2003)
Было предложено множество каталитических подходов для апгрейда и улучшения свойств продуктов быстрого пиролиза. Хорошо известным примером является каталитический крекинг чистой биомассы и/или пиролизного масла до продуктов, не содержащих кислорода. Однако этот подход сопровождается значительным образованием кокса (до 40 мас.% от массы биомассы) (Horne & Williams, 1996; Vispute et al., 2010), что представляет собой серьёзную проблему, требующую решения.
Ряд исследований был направлен на удаление связанного кислорода в форме CO и/или CO₂ путём реакций декарбонилирования и декарбоксилирования, как термических, так и каталитических. Термический процесс известен как HPTT (высокотемпературная обработка при высоком давлении) (De Miguel Mercader et al., 2010). Однако удаление кислорода до уровня ниже 10% представляется затруднительным даже при использовании катализаторов, а получаемый продукт обладает высокой вязкостью, что ограничивает его потенциальное применение.
Более перспективным вариантом считается каталитическая гидрообработка (hydrotreating) или гидропереработка быстрого пиролизного масла (Conti, 1997; Elliott, 2007; Elliott et al., 2009; Kaiser, 1997; Rep et al., 2006). В этом процессе пиролизное масло обрабатывается водородом в присутствии гетерогенного катализатора с целью гидродеоксигенирования, т.е. удаления кислорода и улучшения свойств получаемого продукта.
Одним из потенциально привлекательных направлений использования улучшенных масел является их совместная переработка в существующих нефтеперерабатывающих установках. Это позволит частично заменить ископаемый углерод в жидком моторном топливе возобновляемым углеродом из биомассы, используя уже имеющуюся инфраструктуру, как это предлагается в европейском проекте Biocoup. Такой подход может снизить инвестиционные затраты и преодолеть другие барьеры для внедрения технологии быстрого пиролиза.
Однако сырая пиролизная нефть непригодна для совместной переработки. Она несовместима с типичным нефтяным сырьём (например, вакуумным газойлем) из-за содержания полярных компонентов. Кроме того, она высококислотная (содержание органических кислот достигает 10 мас.%) (Oasmaa, 2003), что вызывает коррозию и может отрицательно повлиять на катализаторы в нефтепереработке (например, цеолиты в FCC-процессе) (Dimitrijevic et al., 2006). Также она склонна к коксованию при повышенных температурах.
Альтернативным направлением использования улучшенного пиролизного масла является прямое применение в качестве зелёного моторного топлива в двигателях внутреннего сгорания. Для этого потребуется глубокая гидроочистка масла до уровня содержания кислорода ниже 1% с целью придания продукту свойств, аналогичных традиционным углеводородным топливам. Однако потребление водорода при этом будет высоким, что негативно скажется на экономике процесса.
Настоящая глава, посвящённая каталитической гидрообработке быстрого пиролизного масла, состоит из нескольких разделов:
- В части 2 приводится краткий обзор типичных катализаторов, используемых для гидрообработки пиролизного масла,
- Далее следует описание основных требований к процессу,
- В части 3 рассматриваются детальные исследования процесса с использованием катализатора Ru/C, включая влияние условий реакции на выход продукта, поглощение водорода и ключевые свойства,
- На основе полученных данных будет представлена сеть реакций,
- В части 5 обсуждаются улучшенные составы катализаторов, их эффективность и оценка производительности.
2. Исследования катализаторов для гидрообработки быстрого пиролизного масла
Хорошо известными катализаторами для гидрообработки пиролизного масла являются традиционные катализаторы гидрообессеривания, такие как NiMo/Al₂O₃, CoMo/Al₂O₃ и NiMo/Al₂O₃–SiO₂ в сульфидированной форме (Ferrari и др., 2002; Maity и др., 2000), поскольку они способны выдерживать жёсткие условия реакции (температура выше 200 °C, водная среда с органическими кислотами).
Также исследовались катализаторы на основе CoMo, поддержанные на углероде, а также молибден на носителях TiO₂, ZrO₂ и их смешанных оксидах TiO₂–ZrO₂ (Satterfield & Yang, 1983; Lee & Ollis, 1984). Все эти катализаторы требуют присутствия серы в сырье, чтобы поддерживать каталитическую активность. Однако в пиролизных маслах содержится лишь небольшое количество серы (Furimsky, 2000), поэтому для поддержания активности катализатора потребуется добавление серы в процессе. Это приведёт к выбросам серы, например, в отходящих газах, что нежелательно с экологической точки зрения.
Катализаторы без серы и без благородных металлов также исследовались, но в значительно меньшей степени. Сообщалось, что Ni/SiO₂ демонстрировал низкую активность по гидродеоксигенированию (HDO) при переработке фенола. Horne & Williams (1996) тестировали цеолиты ZSM-5 в качестве катализатора для удаления кислорода из модельных соединений, таких как анизол. Однако результаты показали, что анизол в основном превращается в фенол и метилзамещённые фенолы, а не в полностью обескисленные соединения.
Xu и др. (2010) протестировали MoNi/γ-Al₂O₃ (предварительно восстановленный) для мягкой гидрообработки пиролизного масла при 3 МПа и 200 °C.
Также проводились исследования катализаторов на основе благородных металлов в качестве замены сульфидных катализаторов. Примеры включают:
- Pd на носителях цеолитного типа (Horne & Williams, 1996),
- Pd на мезопористых CeO₂ и ZrO₂ (Senol и др., 2005),
- Rh на цирконии (Gutierrez и др., 2008),
- Углеродные носители (Wildschut и др., 2009b; 2010a; 2010b; Venderbosch и др., 2010).
Недавно мы представили первичные результаты исследований по апгрейду быстрого пиролизного масла методом каталитической гидрообработки с использованием различных гетерогенных катализаторов на основе благородных металлов (Ru/C, Ru/TiO₂, Ru/Al₂O₃, Pt/C и Pd/C), результаты которых были сравнены с типичными катализаторами гидрообработки, такими как NiMo/Al₂O₃ и CoMo/Al₂O₃ в сульфидированной форме.
Реакции проводились при температурах от 250 до 350 °C и давлениях от 100 до 200 бар. Катализатор Ru/C показал наилучшие результаты по сравнению с классическими катализаторами гидрообработки как по выходу масла (до 60 мас.%), так и по степени обескисления (до 90 мас.%) (см. рисунок 2) (Wildschut и др., 2010).
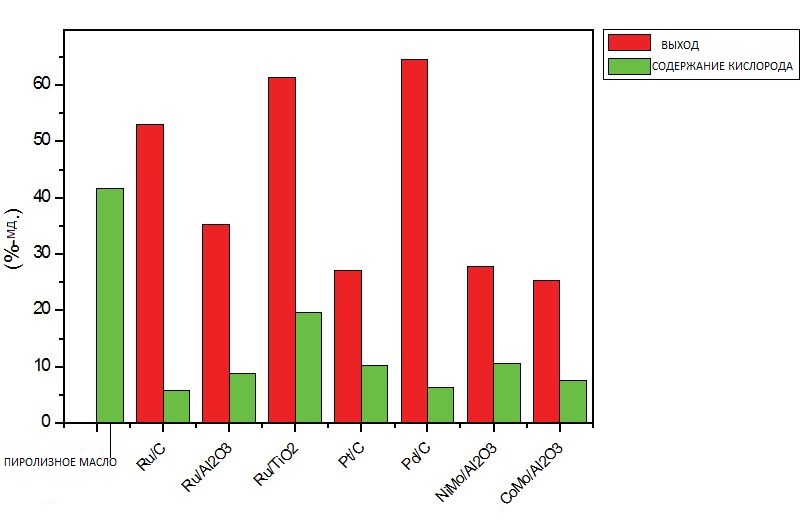
Рисунок 2.
Исследования катализаторов на благородных металлах на различных носителях
К сожалению, катализаторы на основе благородных металлов очень дорогие, что ограничивает их потенциальное применение. Недавно была описана каталитическая гидрообработка быстрого пиролизного масла с использованием значительно более дешёвых биметаллических катализаторов NiCu/δ-Al₂O₃ с различными отношениями Ni/Cu (от 0,32 до 8,1 по массе) при постоянной общей загрузке металлов около 20 мас.% (Ardiyanti и др., 2009).
Реакции гидрообработки пиролизного масла проводились в автоклаве в периодическом режиме: 1 час при 150 °C, затем 3 часа при 350 °C при общем давлении 200 бар. Наибольшая активность катализатора (по поглощению водорода на грамм Ni) была получена для катализатора с наименьшей загрузкой никеля (5,92Ni18,2Cu). Полученные продукты содержали 10–17 мас.% кислорода и демонстрировали улучшенные свойства по сравнению с исходным маслом.
Недавно также была показана эффективность гомогенных катализаторов на основе рутения (Ru) для каталитического апгрейда фракций пиролизного масла (Mahfud и др., 2007a). Фракции выделялись обработкой пиролизного масла дихлорметаном или водой. Обработка дихлорметаном приводила к формированию слоя, обогащённого лигниновой фракцией пиролизного масла. Эта фракция затем подвергалась гидрированию в двухфазной системе (дихлорметан/вода) с применением гомогенного, водорастворимого катализатора Ru/трифенилфосфин-трис-сульфонат (Ru-TPPTS). Анализ показал существенное снижение содержания кислорода, особенно количества реакционноспособных альдегидов.
Водорастворимая фракция пиролизного масла, полученная обработкой масла избытком воды, также гидрировалась в двухфазной системе с использованием гомогенного Ru-катализатора (Ru/трифенилфосфин, Ru-TPP), растворённого в аполярном растворителе (Mahfud и др., 2007b).
Первоначальные эксперименты проводились с модельными соединениями, такими как гидроксиацетальдегид и ацетол. Были изучены и количественно оценены влияния параметров процесса (например, температуры, начального давления H₂ и начальной концентрации субстрата) для ацетола с использованием кинетической модели.
Гидрирование водорастворимой фракции пиролизного масла в двухфазной системе при оптимальных условиях показало значительное снижение количества реакционноспособных альдегидов, таких как гидроксиацетальдегид и ацетол, что свидетельствует о высоком потенциале гомогенных катализаторов Ru для апгрейда пиролизных масел.
3.Исследования процесса каталитической гидрообработки быстрого пиролизного масла
3.1. Введение
Ранее предполагалось, что каталитическая гидрообработка пиролизного масла во многом схожа с традиционными процессами гидрообработки ископаемого сырья (например, гидрообессеривание, гидроденитрификация), и поэтому типичные катализаторы, условия процесса и конфигурация реактора переносились из нефтехимии (Elliott, 2007). Во всех случаях цель — снижение содержания определённых элементов (кислорода, серы, азота) в сырье путём взаимодействия с водородом в присутствии катализатора.
Однако обычные условия реакций, применяемые при переработке продуктов нефти, не могут быть напрямую использованы для пиролизного масла. Например, пиролизное масло нельзя обрабатывать при температуре выше 300 °C, так как оно имеет высокую склонность к закоксовыванию.
Многочисленные публикации по гидрообработке пиролизного масла в насадочных колоннах и автоклавах появились в 1980-х и 1990-х годах, а также совсем недавно (Elliott, 2007). Особенно старая литература носит в основном описательный и экспериментальный характер и фокусируется на простом сборе фактов. В этих работах зафиксированы значительные различия в рабочих условиях, таких как давление, температура, время пребывания и расход водорода.
Во всех случаях, при отсутствии активного катализатора или водорода под высоким давлением, наблюдается интенсивное закоксовывание пиролизного масла.
Снижение образования кокса возможно при проведении каталитической гидрообработки при относительно низкой температуре (175–250 °C), в ходе которой реакционноспособные компоненты масла стабилизируются. Далее стабильный продукт может быть дополнительно обработан при более высоких температурах (> 300 °C) и давлениях (> 150 бар).
Считается, что высокое давление играет ключевую роль в:
- поддержании воды, содержащейся в пиролизном масле, в жидком состоянии (что, вероятно, снижает склонность к коксованию),
- улучшении растворимости водорода в изначально полярном биомасле,
- повышении скорости реакций гидрирования.
3.2. Исследования процесса с использованием катализатора Ru/C
3.2.1. Исследования в периодическом режиме
Недавно мы провели глубокое исследование в периодической установке, чтобы определить влияние условий процесса на каталитическую гидрообработку быстрого пиролизного масла с использованием катализатора Ru/C при температуре 350 °C и давлении 200 бар (Wildschut и др., 2010a).
После реакции жидкий продукт разделялся на три различные фазы:
- слегка жёлтую водную фазу,
- и две масляные фазы коричневого цвета — одна с плотностью выше воды, а другая с плотностью ниже воды.
Кроме того, образовались значительные количества твёрдой фазы (кокс/углерод, около 5 мас.% от массы поданного пиролизного масла) и газообразных органических соединений (CO, CO₂, CH₄).
Выход масла и элементный состав продуктовых фаз показали сильную зависимость от времени реакции. Наибольший выход масла (65 мас.%) был получен после 4 часов реакции при использовании 5 мас.% катализатора от массы пиролизного масла (см. рисунок 3).
При более длительном времени реакции наблюдалось снижение выхода масла за счёт увеличенного образования газообразных компонентов (метан, этан, пропан, CO/CO₂).
Поглощение водорода после 4 часов реакции составило около 400 нм³ на тонну пиролизного масла (в пересчёте на сухое вещество).
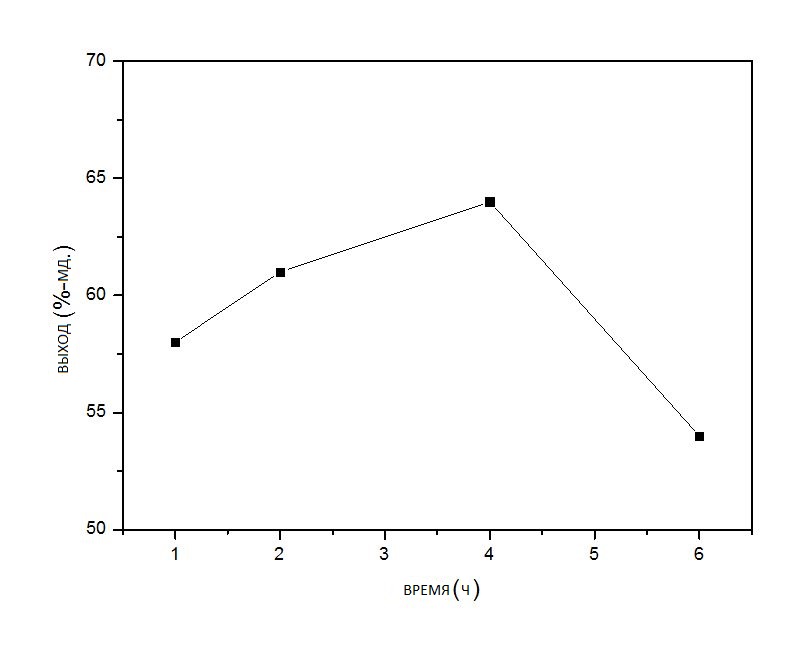
Рисунок 3.
Общий выход масла (в пересчёте на сухое вещество) в зависимости от времени реакции при гидрообработке пиролизного масла
(350 °C, 200 бар, катализатор Ru/C).
Для получения представления о реакционной способности различных классов соединений (фракций) в быстром пиролизном масле в ходе процесса каталитической гидрообработки была использована схема экстракции с использованием двух растворителей, основанная на работе Oasmaa и др. (2003).
Схема фракционирования (см. Рисунок 1) была применена как к исходному пиролизному маслу, так и к продуктам реакции, полученным при различных временах обработки (350 °C и 200 бар).
Результаты приведены на Рисунке 4.
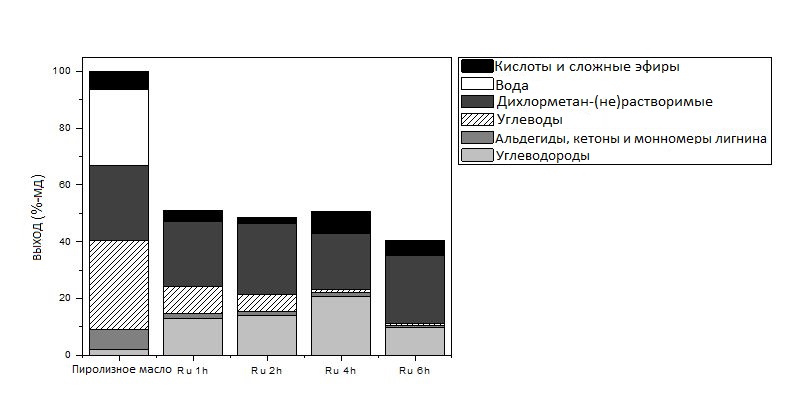
Рисунок 4.
Состав быстрого пиролизного масла и продуктов его гидрообработки (Ru/C, 350 °C, 200 бар) при различных временах реакции, определённый методом экстракции с использованием двух растворителей.
На графике показаны количества различных фракций — углеводов, альдегидов/кетонов/мономеров лигнина, углеводородов, кислот и эфиров — в зависимости от времени реакции.
Чётко виден быстрый спад содержания углеводной фракции со временем. Почти полное превращение углеводов в другие компоненты наблюдается уже через 6 часов, что свидетельствует о высокой реакционной способности этой фракции.
3.2.2. Экспериментальные исследования с Ru/C в непрерывных установках
Недавно были опубликованы глубокие экспериментальные исследования каталитической гидрообработки с использованием катализатора Ru/C в непрерывной установке с насадочными реакторами (Venderbosch и др., 2010). Результаты этого исследования будут приведены далее, так как они дают детальное представление о влиянии условий процесса на выход продуктов, их свойства и протекающие на молекулярном уровне реакции.
Некоторые эксперименты проводились в отсутствие катализатора для изучения чисто термических реакций.
Процесс каталитической гидрообработки осуществлялся в установке, состоящей из четырёх насадочных реакторов, соединённых последовательно. Температуру в каждом реакторе можно было регулировать независимо, что позволяло проводить эксперименты с разными температурными профилями по длине реакционной зоны.
Типичные условия включали:
- давление: от 150 до 300 бар,
- температура: от 150 до 400 °C,
- WHSV (обратное время пребывания): от 2 до 10 кг сырья / кг катализатора в час.
В дальнейшем будут рассмотрены термические реакции, а затем — каталитическая гидрообработка при различных уровнях температуры.
3.2.2.1. Термические реакции
Для детального изучения термических (некаталитических) реакций пиролизное масло пропускалось через реактор без катализатора при давлениях до 300 бар и температурах до 350 °C с временем пребывания от долей секунды до нескольких минут.
Обычно при таких условиях однородное пиролизное масло превращается в:
- вязкую органическую жидкость,
- водную фазу
- и газовую фазу.
Содержание углерода в вязкой фазе составляет около 60 мас.% (по сравнению с 40 мас.% в исходном масле), а содержание кислорода — около 32 мас.%. Образуется дополнительное количество воды — до 30 % от исходного содержания воды в пиролизном масле. Вода распределяется между двумя жидкими фазами, но основная её часть попадает в водную фазу.
С энергетической точки зрения:
- около 80 % тепловой энергии пиролизного масла переходит в вязкий продукт,
- менее 20 % — в водную фазу,
- и около 1 % — в газовую фазу.
Газовая фаза состоит из CO и CO₂ в соотношениях от 1:10 до 1:3 (в зависимости от температуры, давления и времени пребывания), с выходом почти 4 мас.% от поданного пиролизного сырья.
Хотя на молекулярном уровне неясно, какие именно реакции происходят, можно выделить как минимум два параллельных пути:
- Образование газа — в результате декарбоксилирования / декарбонилирования (выделение CO и/или CO₂).
- Обезвоживание — вероятно, через реакции конденсации (полимеризации).
Возможным источником газов являются органические кислоты, содержащиеся в масле. Однако, во всех водных и органических пробах pH остаётся практически таким же, как у исходного пиролизного масла. Это может означать, что:
- либо кислоты не прореагировали,
- либо они одновременно разрушаются и заново образуются.
Подробный анализ кислот в продуктах не проводился, и точный механизм реакций остаётся неясным.
Разбавление пиролизного масла «инертными» растворителями приводит к подавлению реполимеризации. Кроме того, после определённого порога газовыделение перестаёт зависеть от температуры и времени пребывания, тогда как количество образующейся воды продолжает расти. Это говорит о том, что механизм образования газа отличается от механизмов полимеризации.
Разделение фаз при таких условиях может быть вызвано несколькими факторами, например:
- увеличением содержания воды из-за образования воды в реакциях конденсации.
Известно (хотя и не полностью объяснено), что при превышении определённого уровня воды пиролизное масло разделяется на водную и слабо полярную органическую фазы. Также возможной причиной является реполимеризация отдельных молекул/фракций, в результате чего продукты становятся менее растворимыми в воде — например, при преобразовании полярных сахаров, которые изначально играют роль «мостиков» для растворения гидрофильного лигнина (Diebold, 2002).
3.2.2.2. Реакции каталитической гидрообработки
Реакции каталитической гидрообработки проводились при трёх уровнях жёсткости условий процесса:
- Мягкое гидрирование — при температуре 175 или 225 °C.
- Мягкое гидродеоксигенирование (HDO) — при температуре от 225 до 275 °C.
- Глубокое гидродеоксигенирование — при более высоких температурах.
Для глубокого HDO использовались образцы из мягкого HDO, которые предварительно полностью разделялись на фазы. После этого органическая фракция (содержащая около 3 мас.% воды) направлялась на дальнейшую обработку при температуре:
- 350 °C в первых двух сегментах реактора,
- и 400 °C в последних двух сегментах.
3.2.2.2. Реакции каталитической гидрообработки
Реакции каталитической гидрообработки проводились при трёх уровнях жёсткости условий процесса:
- Мягкое гидрирование — при температуре 175 или 225 °C.
- Мягкое гидродеоксигенирование (HDO) — при температуре от 225 до 275 °C.
- Глубокое гидродеоксигенирование — при более высоких температурах.
Для глубокого HDO использовались образцы из мягкого HDO, которые предварительно полностью разделялись на фазы. После этого органическая фракция (содержащая около 3 мас.% воды) направлялась на дальнейшую обработку при температуре:
- 350 °C в первых двух сегментах реактора,
- и 400 °C в последних двух сегментах.

Рисунок 5.
Изображения пиролизного масла (слева), продукта после мягкого HDO (в центре) и после второй стадии HDO (справа)
Содержание кислорода в продуктах зависит от жёсткости условий обработки (см. Рисунок 6 для подробностей). Разделение фаз, происходящее между 175 и 225 °C, приводит к резкому снижению содержания кислорода. Это связано с образованием воды и переходом очень полярных, насыщенных кислородом компонентов в водную фазу. При максимальной жёсткости процесса содержание кислорода в продукте составляет около 15 %, по сравнению с примерно 40 % в исходном пиролизном масле.
Потребление водорода варьируется в пределах от 65 до 250 нм³ на тонну пиролизного масла. Более жёсткие условия приводят к большему поглощению водорода (см. Рисунок 6).
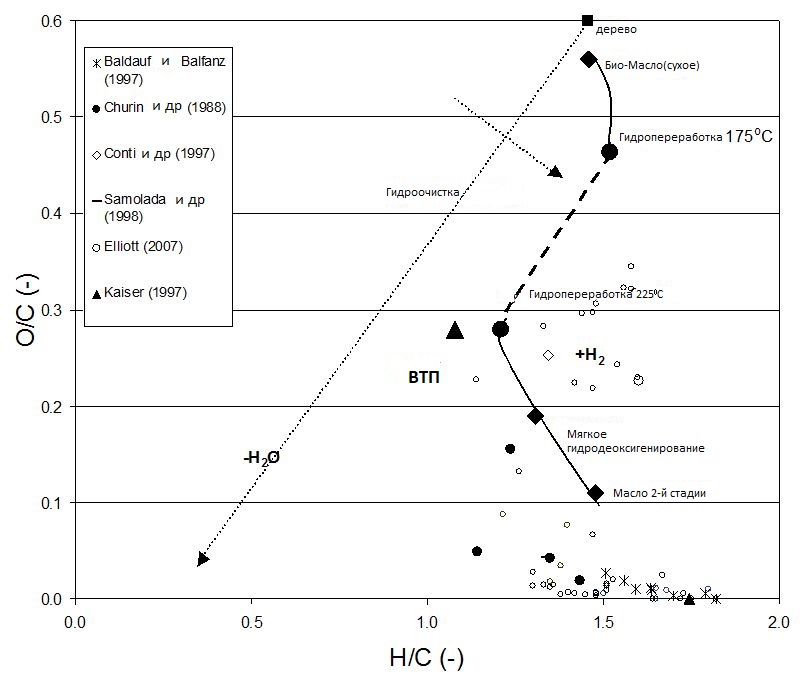
Полезным способом представления изменений в элементном составе продуктов при разных степенях жёсткости процесса является диаграмма ван Крёвелена. В ней отображается соотношение O/C и H/C продуктов в едином графике.
На Рисунке 7 приведён типичный график, составленный на основе литературных данных по гидропереработке пиролизного масла (Elliott, 2007; Venderbosch и др., 2010) и наших результатов с Ru/C при различных жёсткостях. Представлены данные, в том числе для:
- древесины и пиролизного масла,
- четырёх рассмотренных в данной работе случаев: HPTT, гидрообработка при 175 и 225 °C, мягкое HDO и вторая стадия HDO.
Также включены литературные данные из следующих источников: Baldauf и др., 2007; Churin и др., 1988; Conti, 1997; Diebold, 2002; Kaiser, 1997; Samolada и др., 1998. Эти данные получены из различных масел, полученных из разного сырья, обработанных в различных реакторах, с разными катализаторами и при разных условиях.
График также включает:
- кривые, отражающие изменения в элементном составе при гидрообработке,
- теоретическую кривую дегидратации пиролизного масла,
- и тренды для термического пути (HPTT) и гидрообработки, построенные на основе экспериментальных точек.
На основе наших данных по катализаторам Ru/C и подтверждённых точками из литературы на Рисунке 7, можно выделить несколько реакционных путей:
- Реполимеризация пиролизного масла (без катализатора, без водорода, «HPTT»);
- Мягкое гидрирование пиролизного масла (до 250 °C, с катализатором и водородом);
- Обезвоживание масла при температуре около 250–275 °C;
- Гидрообработка пиролизного масла при температуре до 400 °C.
При термической обработке основная реакция — удаление кислорода в виде воды. Также выделяются CO₂ и CO, что немного сдвигает тренд в сторону увеличения отношения H/C (но декарбоксилирование / декарбонилирование ограничено примерно 10 мас.% от сырья). При высокой степени превращения (т.е. при высоких температурах и длительном времени пребывания) образуется обеднённый водородом твёрдый остаток, похожий на традиционный древесный уголь.
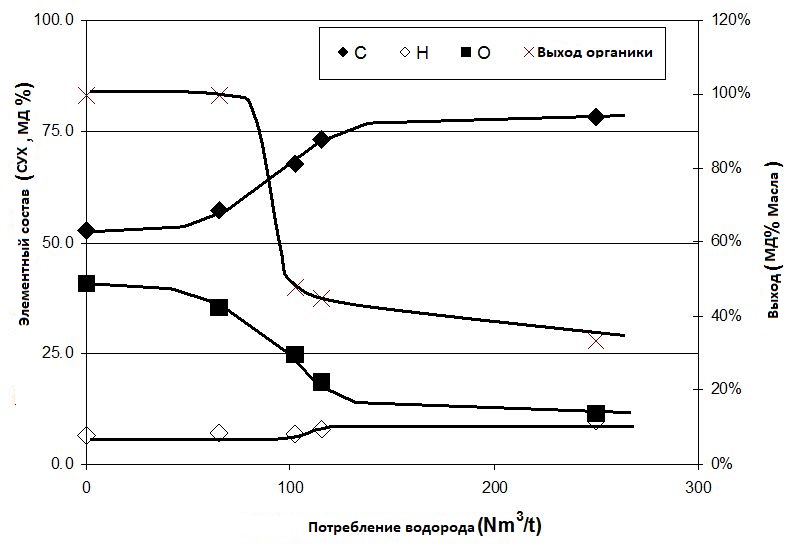
Рисунок 6.
Элементный состав органического масляного продукта (в пересчёте на сухое вещество) в зависимости от потребления водорода для следующих стадий:
- исходное пиролизное масло,
- мягкое гидрирование,
- мягкое гидродеоксигенирование (HDO),
- вторая стадия HDO.
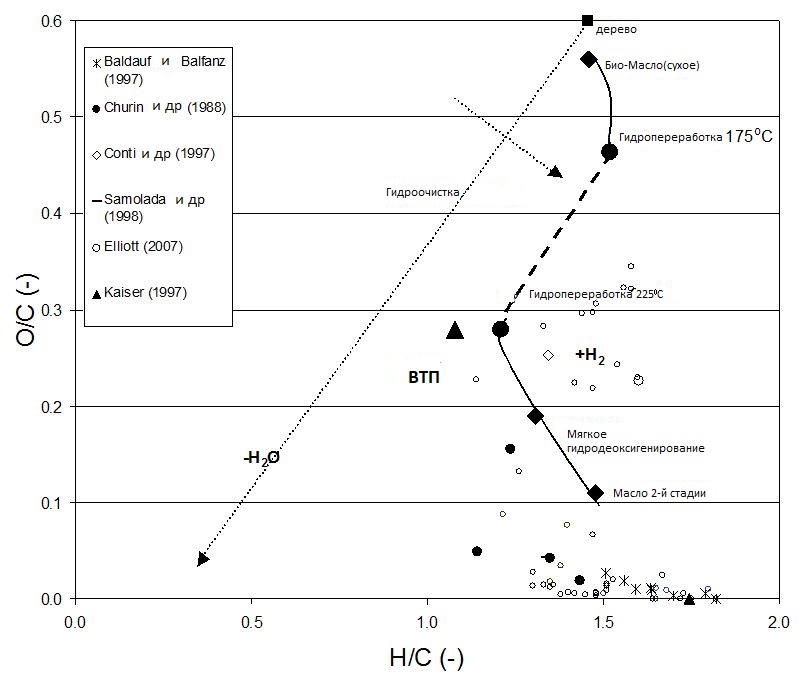
Рисунок 7.
Диаграмма ван Крёвелена для масел, полученных термическим путём (HPTT), после мягкого гидрирования, мягкого HDO и второй стадии HDO, включая соответствующие литературные данные.
Для получения жидкого продукта с более высоким соотношением H/C необходимо дополнительное количество водорода. Этот путь показан на диаграмме:
с последующим гидродеоксигенированием (и, возможно, гидрокрекингом).
он включает мягкую гидрообработку при температуре около 175 °C (без разделения фаз) и 225 °C (с разделением фаз),
3.2.3. Фракционирование продуктового масла: представление о молекулярных изменениях
Различные органические продукты были подвергнуты стандартизированной процедуре жидкостно-жидкостного фракционирования (Oasmaa, 2003, см. Рисунок 1) с целью получить представление о влиянии жёсткости условий гидрообработки на состав продуктов.
Результаты сведены в Рисунке 8 и демонстрируют значительные изменения в составе после проведения реакции.
Исходное пиролизное масло в основном состоит из:
- растворимых в эфире соединений,
- нерастворимых в эфире соединений,
- и воды.
Компоненты этих фракций происходят из целлюлозной и гемицеллюлозной фракций биомассы, причём нерастворимая в эфире фракция особенно богата углеводами.
Содержание растворимых и нерастворимых компонентов, экстрагируемых дихлорметаном (DCM), происходящих из лигниновой фракции биомассы, значительно ниже — всего около 20 % в сумме.
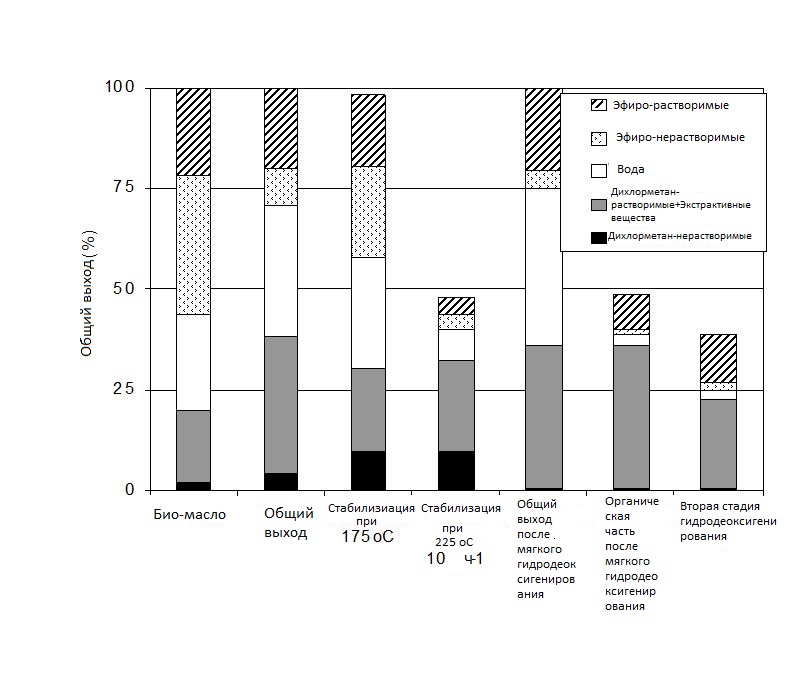
Рисунок 8.
Сравнение результатов фракционирования при различных степенях жёсткости процесса.
3.2.3.1. Термические реакции
При сравнении состава исходного пиролизного масла с продуктами, полученными термическим путём, становится очевидно, что нерастворимые в эфире соединения превращаются в растворимые и нерастворимые в DCM-фракции, а также сопровождаются дополнительным образованием воды.
Аналогичные изменения происходят и с древесными маслами, которые хранились в течение нескольких месяцев или лет — при этом образуются нерастворимые в воде продукты за счёт снижения содержания углеводов (Oasmaa & Kuoppala, 2003).
При высоких температурах и длительном времени пребывания, особенно углеводная фракция становится основной причиной закоксовывания — вероятно, через стадию образования сначала растворимых в DCM веществ, а затем нерастворимых в DCM (т.е. “углеродистых остатков” или «кокса»).
Известно, что при нагревании водных растворов C6-сахаров (например, D-глюкозы, D-маннозы) до температур до 400 °C происходит образование твёрдых веществ. Термическое разложение, как каталитическое (чаще всего под действием кислот), так и некаталитическое, приводит к образованию твёрдых продуктов, известных как гуминовое вещество (гумины) (Girisuta и др., 2006; Watanabe и др., 2005a; 2005b).
Предлагаемый путь реакции включает:
- превращение C6-сахаров в 5-гидроксиметилфурфурол (HMF),
- с последующим образованием левулиновой кислоты (LA) и муравьиной кислоты (FA).
Обе реакции сопровождаются образованием твёрдых гуминовых остатков (см. Схему 1).
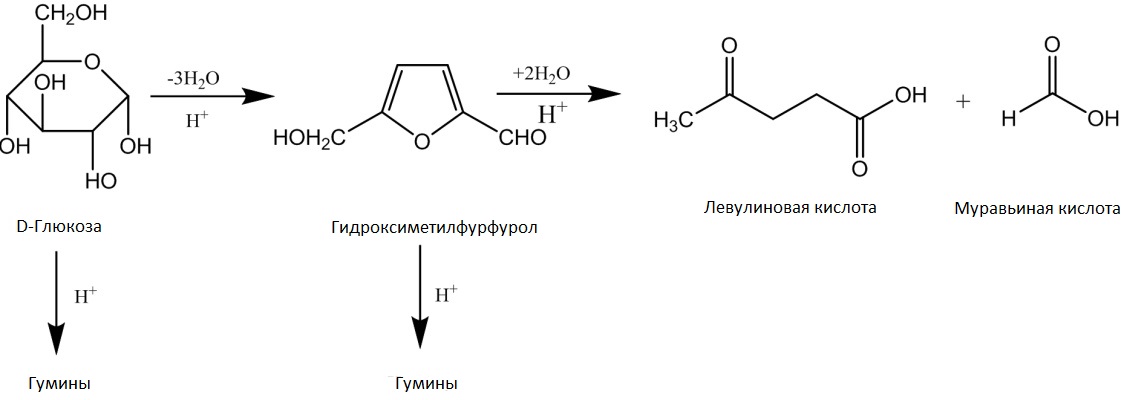
Схема 1.
Реакции разложения D-глюкозы при повышенных температурах.
Повышенные температуры и наличие кислотных катализаторов (как гомогенных, так и гетерогенных) значительно ускоряют разложение D-глюкозы (Girisuta и др., 2006). Подобные реакции могут также происходить и в матрице быстрого пиролизного масла, которое обладает кислотной природой из-за присутствия органических кислот. Эти кислоты катализируют деполимеризацию олигомерных сахаров до D-глюкозы и других C6-сахаров, с последующим превращением в твёрдые остатки, 5-гидроксиметилфурфурол (HMF), а также левулиновую кислоту (LA) и муравьиную кислоту (FA).
Knezevic и др. (2009) исследовали термическое разложение D-глюкозы в горячей сжатой воде при условиях, близких к тем, что используются в каталитической гидрообработке пиролизного масла (240–374 °C). Было показано, что D-глюкоза в основном разлагается до углеродистого остатка (кокса) и некоторого количества газообразных соединений (преимущественно CO₂), в то время как лишь небольшое количество соединений остаётся в водной фазе (например, формальдегид).
При таких условиях реакции протекают очень быстро, и разложение до кокса происходит в течение секунд или нескольких минут.
3.2.3.2. Реакции каталитической гидрообработки
Состав продукта мягкого гидрирования при 175 °C (см. Рисунок 8) значительно отличается от состава исходного пиролизного масла. Количество воды увеличивается незначительно — с 25 до примерно 30 мас.%, чего, по-видимому, недостаточно для разделения фаз. Кроме того, соединения, растворимые в эфире (альдегиды, кетоны, кислоты и т. д.) частично превращаются, но в меньшем объёме по сравнению с процессом HPTT.
Нерастворимая в эфире фракция (углеводы) заметно уменьшается — с 35 до 24 мас.%, тогда как нерастворимая в воде фракция увеличивается пропорционально. Одновременно наблюдается увеличение фракции, нерастворимой в DCM — примерно на 8%, в то время как фракция, растворимая в DCM, увеличивается всего на 3 мас.%.
Подобно процессу HPTT, предполагается, что углеводная фракция в масле частично превращается в более нерастворимые в воде соединения, а также сопровождается дополнительным образованием воды. Однако образующиеся компоненты при мягкой гидрообработке отличаются по характеру от тех, что наблюдаются при HPTT, и в частности, содержание DCM-нерастворимых веществ выше.
Результаты фракционирования продукта, полученного при гидрировании при 225 °C, также приведены на Рисунке 8. Происходит разделение фаз, в результате чего общее количество органической фазы уменьшается. Это приводит к снижению содержания воды, эфирорастворимых и эфиронерастворимых компонентов в органической фазе, что свидетельствует о переносе части веществ в водную фазу.
Рисунок 8 также показывает результат для реакции мягкого HDO. По сравнению с маслами, полученными при более низких температурах, фракция, нерастворимая в DCM, почти полностью превращается в растворимые в DCM компоненты, что говорит о том, что здесь также происходили реакции гидрокрекинга.
При второй стадии гидрообработки наблюдается увеличение содержания эфирорастворимых соединений — за счёт снижения доли растворимых в DCM компонентов и экстрактивных веществ.
3.3. Характеристики продукта
Во всех экспериментах по гидрированию, за исключением тех, что проводились при температуре ниже 200 °C, полученный продукт состоял из двух жидких фаз:
- водной фазы
- и коричнево-красной органической фазы.
Для всех образцов были определены основные характеристики, включая:
- элементный состав (см. выше, Рисунок 7),
- содержание воды,
- и среднемолекулярную массу.
Дополнительно, для оценки склонности к коксованию, образцы анализировали с помощью термогравиметрического анализа (TGA). В этом методе остаточная масса образца, нагретого в атмосфере азота до 900 °C, принималась как показатель склонности к коксованию.
- Высокий остаток указывает на высокую склонность к образованию кокса, а следовательно, на низкую термическую стабильность при повышенных температурах.
Остаточная масса после TGA-анализа сильно зависит от степени жёсткости процесса — подробности см. на Рисунке 9.
Рисунок 9.
Среднемассовая молекулярная масса и остаток после TGA-анализов продуктов, полученных при:
(1) стабилизации,
(2) мягкой гидрообработке,
(3) двухстадийной гидрообработке.
При низкой жёсткости процесса наблюдается увеличение остатка по TGA, при этом максимальное значение (22 %) зафиксировано при средней жёсткости. Дальнейшее повышение жёсткости приводит к резкому снижению остатка по TGA.
Таким образом, можно сделать вывод, что средняя степень жёсткости приводит к образованию продуктов с высоким TGA-остатком, что, в свою очередь, указывает на высокую склонность к коксованию и потенциально меньшую пригодность таких продуктов в качестве сырья для нефтепереработки.
Органические продукты были также проанализированы методом гель-проникающей хроматографии (GPC) для определения среднемолекулярной массы, результаты которой приведены на Рисунке 9.
При мягкой гидрообработке молекулярная масса продуктов возрастает по сравнению с исходным пиролизным маслом, что свидетельствует о протекании реакций полимеризации. Подобный эффект наблюдался и при нагревании пиролизного масла до 275 °C без катализатора (процесс HPTT) (Rep и др., 2006).
При дальнейшем увеличении жёсткости (более высокая температура, меньшие значения WHSV) происходит снижение молекулярной массы, и при максимальной жёсткости она становится менее 300.
Особый интерес представляет взаимосвязь между молекулярной массой продукта и TGA-остатком. Продукты с более высокой молекулярной массой (Mw) также дают более высокий остаток после TGA, что можно объяснить тем, что крупные молекулярные фрагменты служат предшественниками коксующихся соединений.
4. Предлагаемые реакционные пути и их последствия
4.1. Реакционные пути
Схематичное и упрощённое представление предполагаемых реакций, основанное на результатах данной работы, приведено на Рисунке 10.
На начальной стадии процесса гидрообработки происходят каталитическое гидрирование и термическая, некаталитическая реполимеризация, которые протекают параллельно.
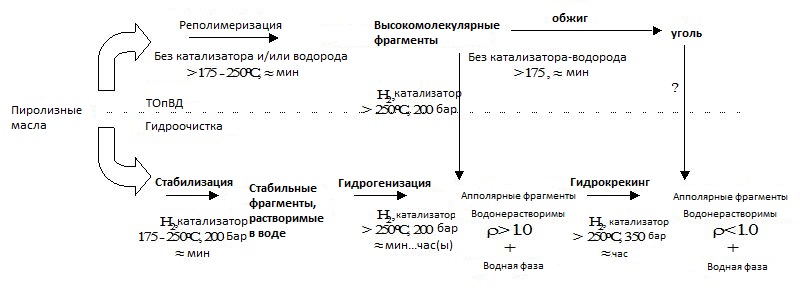
Рисунок 10.
Предлагаемые пути реакций при каталитической гидрообработке пиролизного масла
Реполимеризация приводит к образованию растворимых фрагментов с высокой молекулярной массой, которые при дальнейшем конденсировании превращаются в углеродистый остаток (кокс). Этот путь не является предпочтительным, и скорость полимеризационных реакций должна быть максимально снижена.
Предпочтительный путь включает гидрирование термически нестабильных компонентов в пиролизном масле с образованием стабильных молекул, которые не склонны к полимеризации. Последующие реакции (гидрирование и гидрокрекинг), происходящие на шкале времени в несколько часов, приводят к продуктам с пониженным содержанием кислорода и, в конечном счёте, к увеличению соотношения H/C (см. Рисунок 7).
Наблюдаемое увеличение молекулярной массы органической фазы при низкой жёсткости обработки (см. Рисунок 9) указывает на то, что при использовании катализатора Ru/C реполимеризации избежать невозможно. Однако при более жёстких условиях наблюдается снижение среднемолекулярной массы, что говорит о возможной деполимеризации растворимых тяжёлых фрагментов под действием водорода и катализатора.
Как уже отмечалось, пиролизное масло содержит большое количество олиго- и мономерных сахаров, происходящих из целлюлозной и гемицеллюлозной фракций лигноцеллюлозной биомассы. В этом контексте важно сравнить предложенные пути реакций для пиролизного масла с типичными реакциями гидрирования и термического разложения углеводов при различных условиях.
Термическое разложение мономерных сахаров в водной среде хорошо изучено и известно, что оно приводит к олигомеризации, затем к нерастворимым гуминовым веществам (Girisuta и др., 2006). Например, Knezevic и др. (2009) исследовали разложение D-глюкозы в горячей сжатой воде при 240–374 °C. Результаты показали образование твёрдых веществ (кокса, гуминов), а также небольшого количества газов (преимущественно CO₂). При таких условиях реакции происходят очень быстро, и образование кокса происходит за секунды или минуты.
Каталитическая гидрообработка углеводов с использованием гетерогенных катализаторов широко описана в литературе. Основное внимание уделяется гидрированию D-глюкозы в D-сорбит, известный химикат, применяемый в фармацевтике и пищевой промышленности (Kusserow и др., 2003).
Гидрообработка D-глюкозы на катализаторах на основе Ni, Ru и Pd при 80 °C и 80 бар приводит к получению D-сорбита с высоким выходом (Crezee и др., 2003; Makkee и др., 1985) (см. Схему 2).
Реакции гидрирования при таких низких температурах можно рассматривать как стадию стабилизации в процессе апгрейда пиролизного масла.
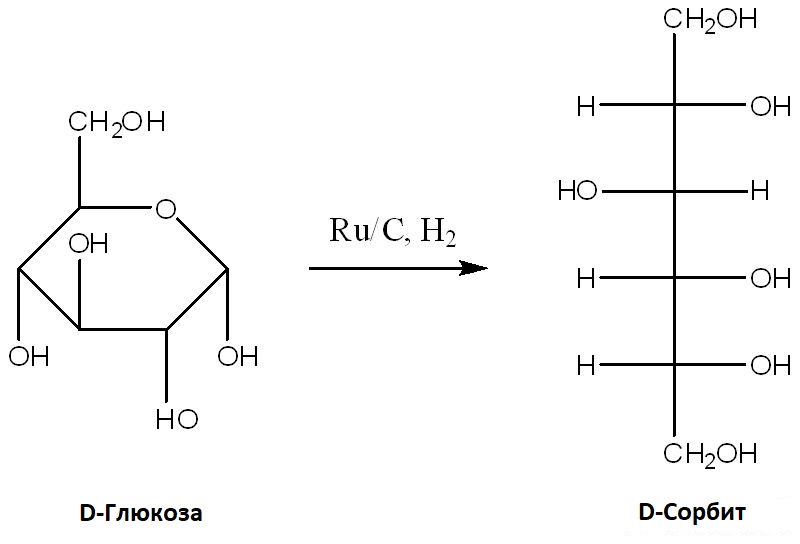
Схема 2.
Каталитическое гидрирование D-глюкозы в D-сорбит
В присутствии водорода и катализатора, D-сорбит не является инертным при повышенных температурах (выше 180 °C) и может превращаться в разнообразные продукты.
Например, Huber и др. (2004) показали, что D-сорбит можно преобразовать в n-гексан с высоким выходом с использованием катализаторов Pd или Pt на SiO₂ или Al₂O₃ при 225–265 °C и давлениях 26–58 бар.
На катализаторе Ru/SiO₂ гидрогенолиз D-соргита при 180–240 °C и 80–125 бар водородного давления даёт преимущественно глицерин и 1,2-пропандиол (Sohounloue и др., 1982) (см. Схему 3). Это указывает на то, что разрыв C–C связей происходит легко, приводя к образованию соединений с меньшей молекулярной массой.
Скорее всего, аналогичные реакции также происходят при каталитической гидрообработке быстрого пиролизного масла, что объясняет образование менее полярных продуктов с низкой молекулярной массой при более жёстких условиях процесса.
Таким образом, можно сделать вывод, что типичные пути реакций пиролизного масла при мягких условиях гидрообработки схожи с реакциями низкомолекулярных сахаров, а именно:
- реполимеризация до твёрдых гуминовых веществ,
- гидрирование и разрыв C–C связей с образованием, например, полиолов,
- и в конечном итоге — углеводородов.
Это подтверждает нашу первоначальную гипотезу, что пиролизное масло следует рассматривать как «сироп, богатый углеводами», а не как традиционную углеводородную жидкость ископаемого происхождения.
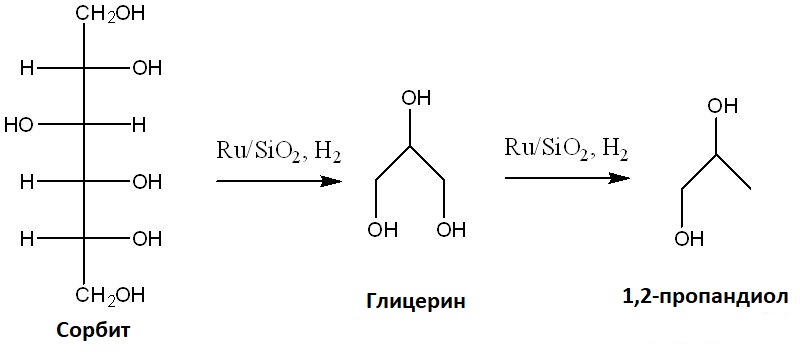
Схема 3.
Гидрогенолиз D-соргита до глицерина и 1,2-пропандиола
4.2. Последствия для технологии
Предложенный путь реакций при каталитической гидрообработке пиролизного масла (см. Рисунок 10) предполагает, что скорость реакции гидрирования должна быть значительно выше, чем скорость реполимеризации, чтобы получить качественное улучшенное пиролизное масло с:
- низкой молекулярной массой,
- низкой вязкостью,
- низкой склонностью к коксованию.
Очевидное решение — это разработка высокоактивных катализаторов гидрирования. Эти исследования будут описаны в следующем разделе статьи.
Однако также следует учитывать грамотный выбор условий процесса и конфигурации реактора, особенно с целью усиления путей гидрирования/гидродеоксигенирования по сравнению с путём реполимеризации. В этом контексте важно хотя бы качественно понимать факторы, определяющие скорость каждого из путей (гидрирование против реполимеризации).
Схематичный график представлен на Рисунке 11, где по оси абсцисс отложена температура, а по оси ординат — предполагаемая скорость реакции (в условных единицах, моль реагента/мин).
На графике представлены три случая:
- Скорость реакции ограничивается переносом водорода из газа в жидкость,
- Доминируют реакции каталитической гидрообработки,
- Преобладают полимеризационные реакции.
Рисунок 11 построен на основе упрощённой кинетики реакций гидрирования и полимеризации глюкозы, однако подробное описание всех допущений выходит за рамки данного раздела, поэтому точные значения на осях x и y опущены.
В расчёте графика учитывались следующие зависимости:
- Скорость гидропроцессов RHR_HRH (моль/м³ реакционного объёма в секунду) можно выразить как произведение внутренней кинетической константы kRk_RkR и удельной площади поверхности катализатора на объём реактора. Поскольку это каталитическая реакция, влияние температуры здесь может быть очень значительным.
- Общий перенос водорода от газа к твёрдой фазе (массообмен) зависит от геометрии реактора и эксплуатационных параметров.
- В реакторах с мешалкой (включая автоклавы) важна скорость перемешивания,
- В насадочных колоннах — смачиваемость частиц катализатора.
В обоих случаях ключевыми факторами являются: - концентрация водорода (т.е. его давление),
- размер частиц катализатора,
- и в меньшей степени — температура.
- Скорость полимеризации RpR_pRp главным образом зависит от температуры и, поскольку это реакция порядка > 1 (возможно 2 или 3), — от концентрации исходного вещества.
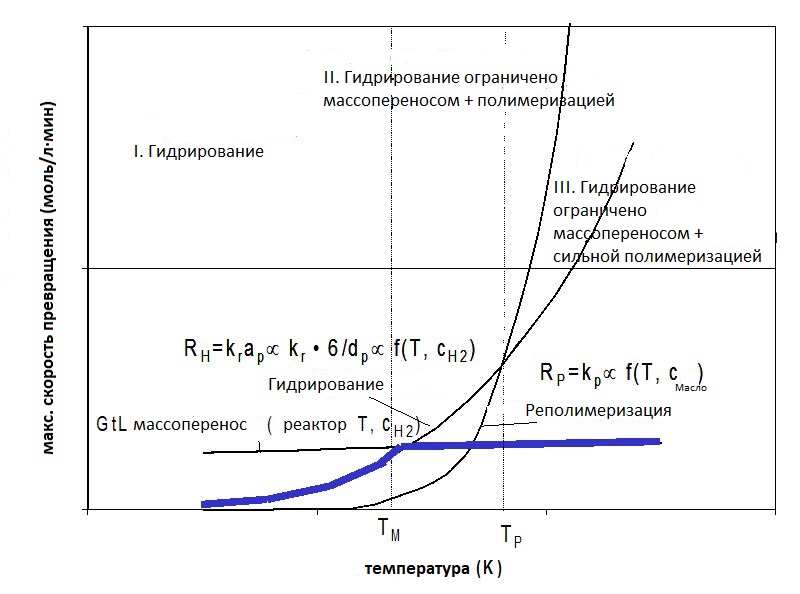
Рисунок 11.
Схематическое представление скоростей превращения в зависимости от температуры для трёх процессов:
- массообмена (газ–жидкость),
- гидрирования,
- полимеризации.
Синяя сплошная линия обозначает суммарную эффективную скорость превращения.
Рассматриваются несколько вариантов для усиления пути гидрирования:
или увеличение разности концентраций водорода между газовой и жидкой фазой.
Увеличить скорость реакций гидропереработки, например:
за счёт повышения загрузки катализатора,
или за счёт увеличения эффективной концентрации водорода в жидкой фазе (через повышение давления, использование растворителя с высокой растворимостью водорода и т. д.).
Снизить скорость полимеризационных реакций, в том числе путём:
выполнения стадии начальной стабилизации при низкой температуре (< 100 °C),
снижения концентрации реагентов (например, путём разбавления).
Увеличить общий коэффициент массообмена газ–жидкость, если реакция ограничивается именно этой стадией.
Это можно реализовать через:
увеличение площади межфазного контакта в реакторе,
повышение коэффициента массообмена,
5. Улучшенные формулы катализаторов для каталитической гидрообработки быстрого пиролизного масла
Разработка высокоактивных металлических катализаторов имеет ключевое значение для снижения склонности к реполимеризации во время каталитической гидрообработки. Все данные, представленные в этой главе до настоящего момента, основаны на использовании катализатора Ru/C. Однако рутений — это дорогой благородный металл, и существует интерес не только к более активным, но и более доступным по цене катализаторам для реакций гидрообработки.
Одним из решений является использование дешёвых биметаллических катализаторов на основе никеля (Ni). Никель известен своей высокой активностью в реакциях гидрирования широкого круга органических функциональных групп, особенно кетонов и альдегидов, и поэтому он рассматривается как потенциально активный металл для гидрообработки.
Тем не менее, монометаллические катализаторы на основе никеля (на носителях из кремнезёма, γ- или δ-оксида алюминия и др.) не подходят для использования в качестве катализаторов гидрирования при типичных температурно-давленостных условиях, применяемых в этих исследованиях. Основных причин — две:
- Никель требует высоких температур восстановления (обычно около 700 °C) для полной активации.
- Катализаторы на основе никеля склонны к быстрой дезактивации при высоких температурах из-за отложения углерода («коксования»).
Отложение углерода может блокировать поверхность никеля или входы в поры носителя, что приводит к снижению скорости реакции.
Обе эти проблемы были решены, в частности, путём введения дополнительного элемента (металлического или неметаллического) — так называемого промотора. Один из таких промотированных катализаторов был детально изучен и далее будет обозначаться как катализатор D.
Рисунок 12 демонстрирует жидкие продукты после гидрообработки с использованием катализатора D при различной степени жёсткости процесса. Слева показано исходное масло, справа — масло, полученное при самых жёстких условиях обработки в данном исследовании.
Интересно, что продукты, полученные на катализаторе D, выглядят намного более прозрачными, чем те, что были получены с использованием катализатора Ru/C.

Рисунок 12.
Внешний вид жидкой фазы после гидрообработки с использованием катализатора D
Диаграмма ван Крёвелена даёт ценную информацию о различиях в производительности между катализатором D и Ru/C (см. Рисунок 13). Для обоих катализаторов наблюдается сходный характер изменений в зависимости от жёсткости процесса, однако кривая для катализатора D смещена к более высоким значениям отношения H/C.
Таким образом, при одинаковом содержании кислорода, отношение H/C выше для катализатора D, что свидетельствует о более высокой скорости гидрирования, а это, как известно, благоприятно сказывается на свойствах продукта.
Реакции реполимеризации, по-видимому, протекают в меньшей степени при использовании катализатора D, чем при применении Ru/C. Это видно из сравнения среднемолекулярной массы конечных продуктов (см. Рисунок 14a), полученных методом гель-проникающей хроматографии (GPC).
- Для Ru/C среднемолекулярная масса значительно увеличивается — с 400 до 1000 Да при низкой жёсткости.
- В то время как для катализатора D она остаётся практически постоянной, в пределах 400–450 Да по всему диапазону содержания кислорода.
Остатки после TGA-анализа масел, полученных с использованием катализатора D (Рисунок 14b), показывают, что углеродистый остаток составляет около 5 %.
Удивительно, но уже при менее жёстких условиях, в отличие от испытаний с другими катализаторами, достигается значительное снижение остатка по TGA.
Таким образом, при использовании катализатора D удаётся получить продукты с более высоким соотношением H/C и меньшим углеродистым остатком, что говорит о том, что скорости реакций гидрирования/гидродеоксигенирования на катализаторе D выше, чем на Ru/C.
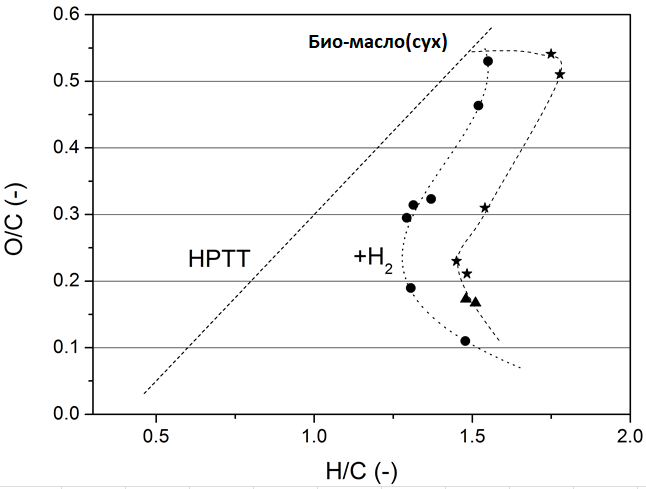
Рисунок 13.
Диаграмма ван Крёвелена для масел, полученных с использованием катализатора Ru/C (круги) и катализатора D (штриховка). Линии на графике — тренды.
Одним из важнейших свойств улучшенных масел является их вязкость. На Рисунке 15 представлено сравнение профиля вязкости масел в зависимости от содержания кислорода для традиционных катализаторов (Ru/C, NiMo, CoMo) и катализатора D.
Явно видно, что при среднем содержании кислорода вязкость у катализатора D значительно ниже. Хотя для полного анализа необходимо провести испытания и при крайне низком содержании кислорода, уже очевидно, что катализатор D обеспечивает получение продуктов с более низкой вязкостью, чем традиционные катализаторы.
Низкая вязкость, скорее всего, обусловлена:
- меньшей средней молекулярной массой продуктов (что подтверждено ранее),
- и высокой скоростью реакций гидрирования/гидродеоксигенирования на катализаторе D по сравнению с Ru/C.
Основываясь на свойствах масел, полученных с использованием катализатора D, можно сделать вывод, что реполимеризация практически не происходит. В результате удаётся получить:
- масла с меньшей молекулярной массой,
- меньшей вязкостью,
- и меньшим остатком по TGA.
Таким образом, реакционный путь для катализатора D можно значительно упростить — см. Рисунок 16 для наглядности.
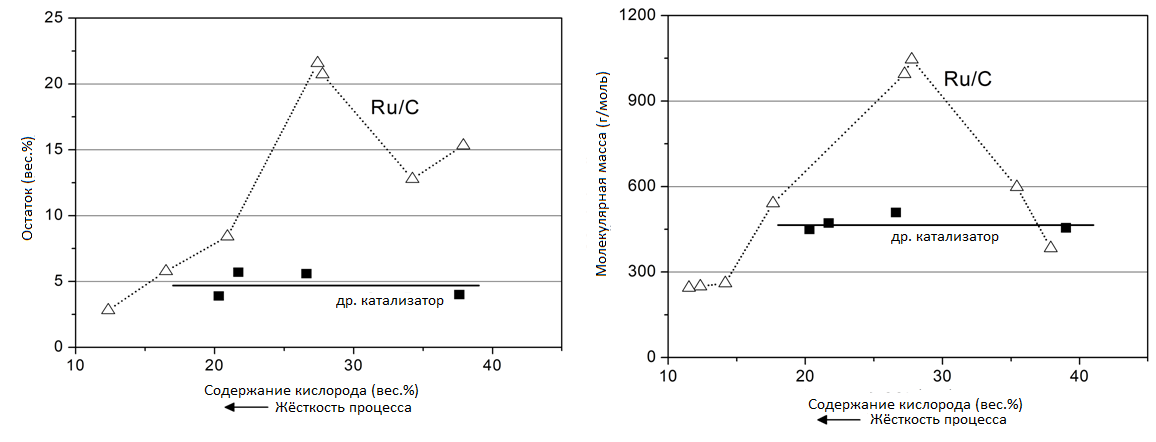
Рисунок 14.
Остаток после термогравиметрического анализа (TGA, в мас. %) для катализаторов Ru/C и катализатора D (вверху);
Среднемолекулярная масса конечного продукта (внизу).
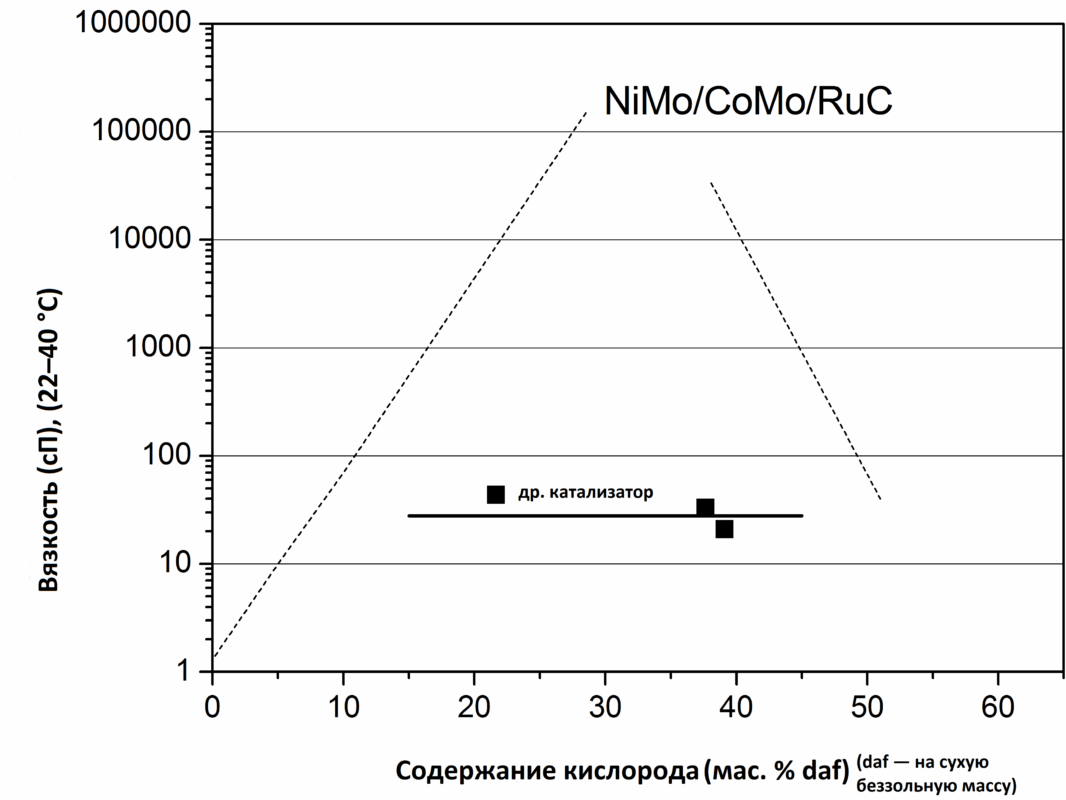
Рисунок 15.
Вязкость масла в зависимости от содержания кислорода для традиционных катализаторов и катализатора D.

Рисунок 16.
Предлагаемые пути реакций при мягкой гидрообработке пиролизных масел с использованием катализатора D.
6.Потенциал применения улучшенных масел и критические свойства продукта
Цель описанного в данной работе исследования — получение продуктов на основе пиролизного масла, пригодных для совместной переработки в нефтеперерабатывающих заводах. Важно обсудить некоторые ключевые свойства продуктов, которые, вероятно, играют решающую роль для такого применения.
Одним из важнейших показателей является склонность масла к образованию кокса при нагревании, которая может быть определена по:
- методу остатка Конрадсона (CCR),
- или по остатку после термогравиметрического анализа (TGA) (см. выше) (Furimsky, 2000).
В целом, пиролизные масла показывают значения CCR на уровне 20–30 % (Samolada, 1998), что ограничивает их прямое применение в качестве совместного сырья.
TGA-остатки для улучшенных масел, полученных с использованием Ru/C и катализатора D, варьируются от 3 до 22 мас.% (см. Рисунок 14) и могут регулироваться жёсткостью процесса (температурой, временем пребывания или партией).
Важно отметить, что низкие значения TGA-остатков возможны даже для продуктов, содержащих значительное количество связанного кислорода (> 10 мас.%). Это означает, что можно получить стабильные масла даже при относительно высоком содержании кислорода.
С технологической точки зрения это также преимущество, поскольку позволяет сократить потребление водорода в процессе каталитической гидрообработки — одного из основных переменных затрат.
Недавно были проведены исследования по совместной переработке гидрообработанных пиролизных масел, полученных с использованием катализатора Ru/C (с содержанием кислорода > 5 мас.%) в лабораторной модели установки каталитического крекинга (FCC, MAT) (de Miguel Mercader и др., 2010).
Гидрообработанные продукты успешно растворялись и перерабатывались совместно с Long Residue (20 мас.% улучшенного масла).
Выход бензина FCC составил 44–46 мас.%, а лёгкого циклического масла (LCO) — 23–25 мас.%, что близко к показателям базового сырья, при этом чрезмерного увеличения кокса и сухого газа не наблюдалось.
Однако при использовании неразбавленного улучшенного масла результаты были менее успешными — выходы кокса и сухого газа значительно увеличивались по сравнению с со-подачей. Это ясно демонстрирует необходимость совместной переработки для достижения хороших показателей.
Это исследование также показало, что, вопреки первоначальным предположениям, необязательно добиваться содержания кислорода ниже 1 мас.% в улучшенном масле.
В настоящее время продолжаются испытания MAT с продуктами, полученными с катализатором D, что позволит установить детальные зависимости «процесс — свойства продукта» для совместного использования с нефтяным сырьём.
Улучшенные масла, полученные с катализатором D, обладают значительно более высокой термической стабильностью, чем исходное пиролизное масло, что подтверждается низкими TGA-остатками (см. Рисунок 14).
Предварительные исследования показали, что это позволяет перегонять масло на фракции без образования чрезмерного количества кокса.
Дальнейшие детальные исследования находятся в процессе и будут опубликованы в ближайшее время.
7.Выводы
Апгрейд пиролизного масла с помощью каталитической гидрообработки на гетерогенных катализаторах был подробно исследован с использованием катализатора Ru/C.
Исследования дали ценную информацию о химических превращениях, происходящих в процессе гидрообработки, включая как термические, так и гидрирующие/гидродеоксигенирующие пути.
Путь реполимеризации, при котором масла конденсируются в растворимые олигомеры, а затем в углеродистые остатки (кокс), конкурирует с реакциями каталитического гидрирования.
Если в системе присутствует водород и подходящий катализатор, эти растворимые олигомеры могут быть деполимеризованы до стабильных компонентов, которые в дальнейшем можно апгрейдировать.
С этой точки зрения пиролизные масла проявляют реакционную способность, типичную для углеводов, что объясняется высоким содержанием олиго- и мономерных сахаров в составе масла.
Новые, высокоактивные катализаторы на основе никеля (Ni) были разработаны и показали значительно лучшую эффективность, чем традиционные катализаторы, обеспечивая продукты с улучшенными свойствами.
В будущем планируется проведение дополнительных экспериментов с целью:
- уточнения реакционных путей при каталитической гидрообработке,
- и разработки эффективных процессов получения стабильного масла с желаемыми свойствами при минимальных производственных затратах.
Задачи включают:
- Оценку влияния конфигурации реактора на скорости реакций (включая проблемы массообмена) и выбор оптимального типа реактора;
- Определение релевантных физических свойств, например растворимости водорода;
- Оптимизацию условий гидрообработки, особенно с учётом необходимого уровня потребления водорода;
- Анализ взаимосвязи «продукт — процесс»: необходимо чётко определить степень жёсткости процесса, требуемую для последующей совместной переработки на НПЗ;
- Исследование источников и доступности водорода — возможно, применим даже синтез-газ;
- Оценку влияния экзотермичности реакции, которая также требует уточнения.